This is the first reported case of siblings with Solitary Median Maxillary Central Incisor (SMMCI) syndrome presenting with an erupted single symmetrical central maxillary midline incisor between normal central incisors. Usually only one central incisor is present. This case is also interesting as the tooth described could have been mistaken for a mesiodens, however, a mesiodens is not symmetrical and usually erupts between the maxillary central incisors and sits slightly to the right or left of the midline.
CPD/Clinical Relevance: This case highlights the fact that SMMCI should not be considered as a simple dental anomaly because it may be associated with more complex craniofacial malformations.
Article
Hannah Morison
Solitary Median Maxillary Central Incisor (SMMCI) syndrome is a complex disorder and a rare dental anomaly.1 SMMCI occurs more in women and prevalence is estimated at 1:50,000 live births, with a higher incidence in stillbirths and aborted foetuses. The aetiology is uncertain, but is thought to be associated with a missense mutation in the Sonic HedgeHog (SHH) gene.2 It is a unique developmental abnormality arising from an unknown event or events occurring between the 36—38th days in utero.3
SMMCI was initially described as an isolated defect, but has since been associated with multiple, mainly midline defects and HoloProsEncephaly (HPE). HPE is a developmental defect of the forebrain where the cerebral hemispheres fail to separate into distinct halves, leading to midline defects.1 These include defects of the cranial bones, the maxilla, nasal bones, facial bones, the dentition and the brain. It can also cause defects in other midline structures of the body and can be associated with short stature.1,2
The SMMCI tooth is unique and has three distinct features:
A symmetrical crown and root with the crown and root size the same as that of a normal central incisor;4
It develops precisely in the midline of the maxilla; and
It occurs in the primary and secondary dentition.1
It is thought that this is the first reported presentation of a family with SMMCI syndrome where the midline central incisor is a supernumerary/supplemental tooth in the presence of a normal complement of incisors. This case is interesting as the tooth described could have been mistaken for a mesiodens tooth, but a mesiodens is usually asymmetric and, although it erupts approximately in the midline between the central incisor teeth, it always sits slightly to either the right or left of the midline.1
Female – 7 years 4 months
This female was born pre-term at 36 weeks by emergency caesarean section due to foetal tachycardia. Several complications were reported such as low birth weight due to Intra-Uterine Growth Restriction (IUGR), hypoglycaemia, respiratory distress and septicaemia. Her hypoglycaemia was thought to be due to significant IUGR as her birth weight and OccipitoFrontal head Circumference (OFC) were below third centile.
When she was 2 months, it was noted that she had very noisy breathing. On examination, it was reported that she was very snorty and snuffly with a very small nose, but with a normal chest. She was referred to the Ear Nose and Throat (ENT) team as there had been a history of difficulty passing a nasogastric tube soon after birth, where only one of her nostrils was able to have a tube passed. A Computed Tomography (CT) scan revealed possible right-sided choanal atresia. It was later confirmed that there was no evidence of choanal atresia, but narrowing of the choanal passages. The ENT team reported a slight unusual prominence at the end of her nose, therefore Magnetic Resonance Imaging (MRI) was carried out. This showed that she had a dermoid type cyst extending from the tip of her nose up to her foramen caecum, but not extending intracranially. The MRI also showed an incidental finding of schizencephaly in the right lobe of the brain and a referral to neurology was made. The scans also showed that she had three central incisors in her maxilla and a referral to the dental team was made.
Following the referrals, she was confirmed to have nasal dermoid, pyriform aperture stenosis, right frontal schizencephaly and Solitary Median Maxillary Central Incisor (SMMCI) syndrome.
When she was 2 years old she sustained a small uncomplicated enamel fracture to her upper midline central incisor tooth. As she was asymptomatic, her parents opted to leave the tooth in situ, with the decision to extract this tooth in a joint General Anaesthetic (GA) should a GA be planned in the future.
An open rhinoplasty approach was subsequently taken for the full excision of the nasal dermoid, together with the extraction of the midline central incisor under GA at age 3. At 5 years of age, there was a complete deciduous dentition with a maxillary midline diastema following extraction of the midline central incisor.
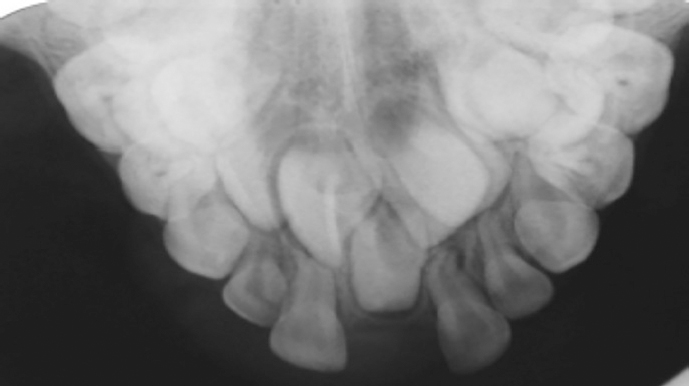
Radiographs and photographs were taken at 6 years old which revealed an unerupted supernumerary symmetrical midline central incisor, together with a full complement of permanent teeth (Figures 1 and 2).
Figure 1. Intra-oral view of female at 6 years old.Figure 2. Upper anterior occlusal radiograph of female at 6 years old.
It was determined that surgical removal of the supernumerary would be needed if it failed to erupt.
The upper anterior occlusal radiograph (Figure 2) showed:
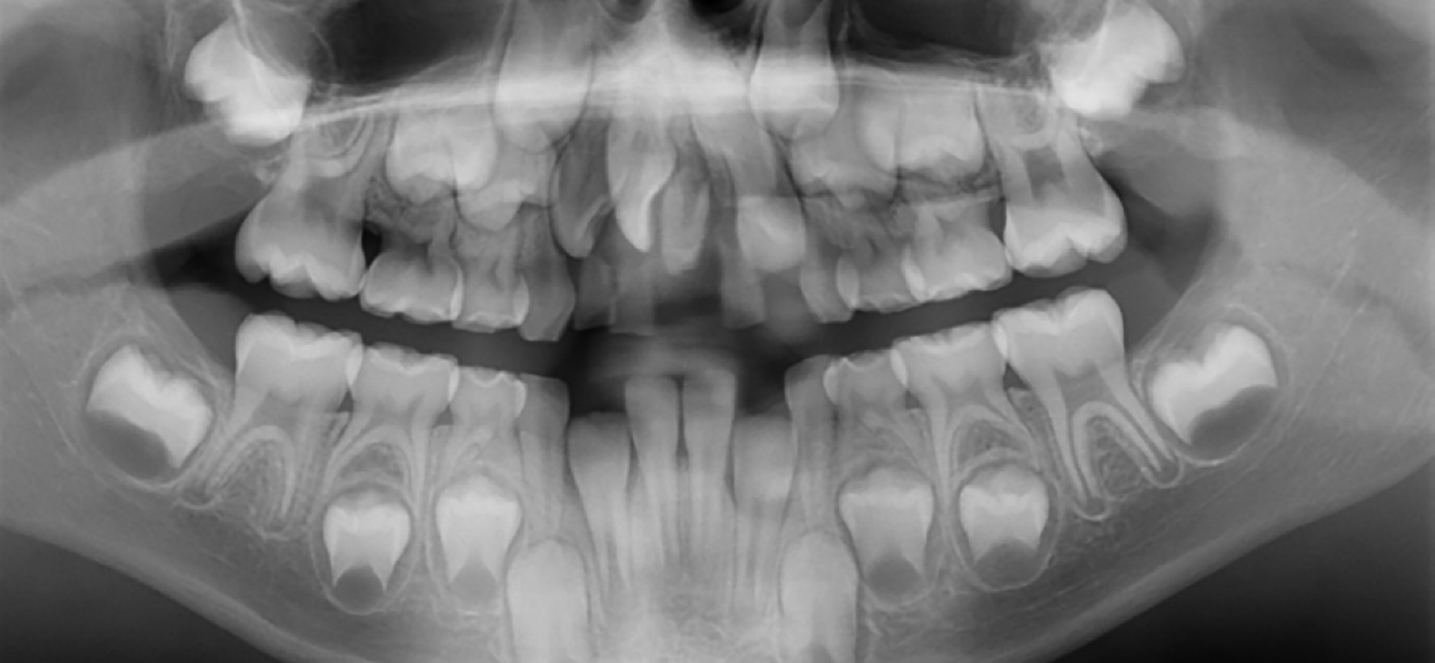
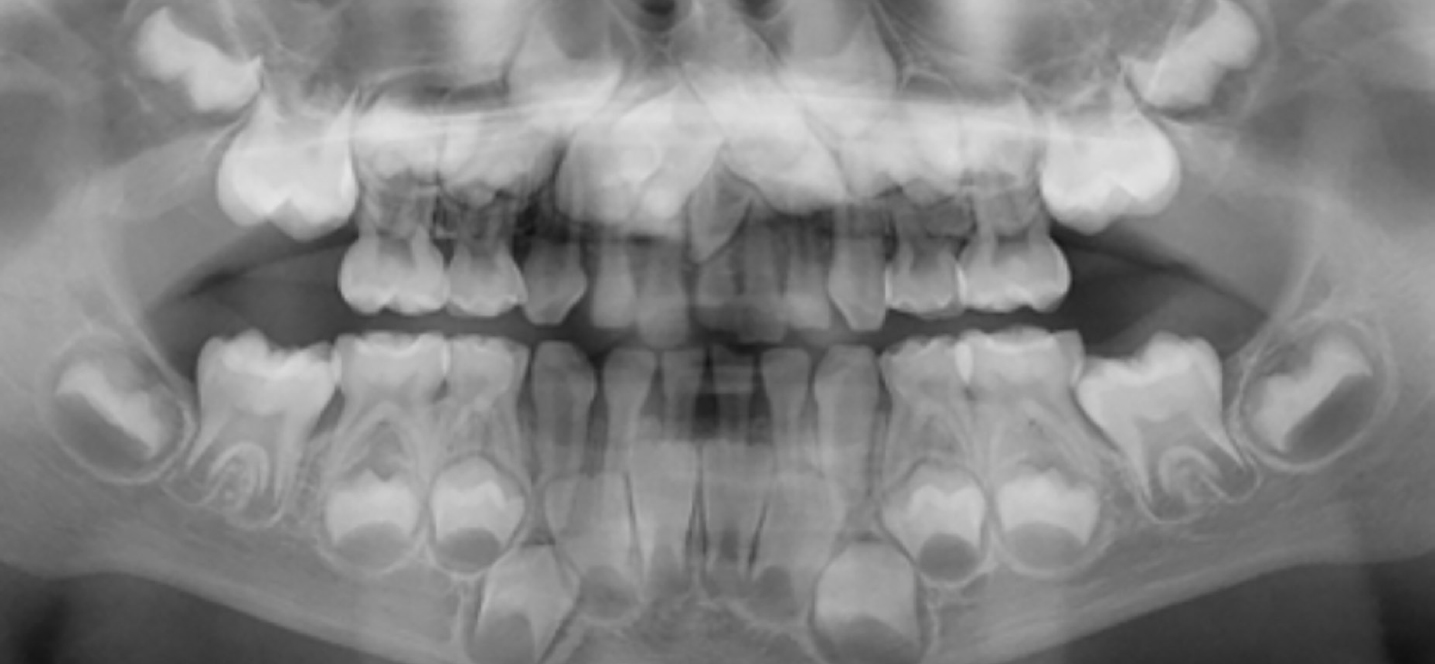
An orthopantomogram (OPG) was taken when she was 7 years old (Figure 3) which identified all her permanent dentition was present apart from her wisdom teeth and, in addition, the presence of a symmetrical midline supernumerary tooth.
Figure 3. Orthopantomogram of female at 7 years old.
Male – 5 years 1 month
This male was born at 40 weeks by Spontaneous Vaginal Delivery (SVD). He had several complications following birth, including Intra-Uterine Growth Restriction (IUGR) and bilateral pneumothorax, for which he was admitted to the Paediatric Intensive Care Unit (PICU) and ventilated for 4 days. At 9 months of age, he attended the Dental Department and it was confirmed that he had an additional central incisor, like his sister. Dental options were given at this stage, which included monitoring or extraction of the upper midline central incisor tooth, which was in addition to normal central and lateral incisors. The option taken was to monitor the tooth with a review planned in 6 months.
An MRI had been carried out in the neonatal period which revealed a normal brain (no schizencephaly), a nasal dermoid cyst and confirmed an obvious midline incisor. The child was therefore referred to ENT, genetics and cardiology. Further imaging showed that he had an enlarged foramen caecum and a frontonasal dermoid cyst with transcranial extension. He was therefore on the mild end of the HPE spectrum. The frontonasal dermoid cyst with transcranial extension was excised with an anterior fossa repair at age 3.
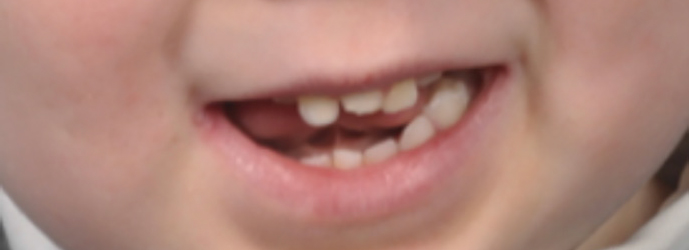
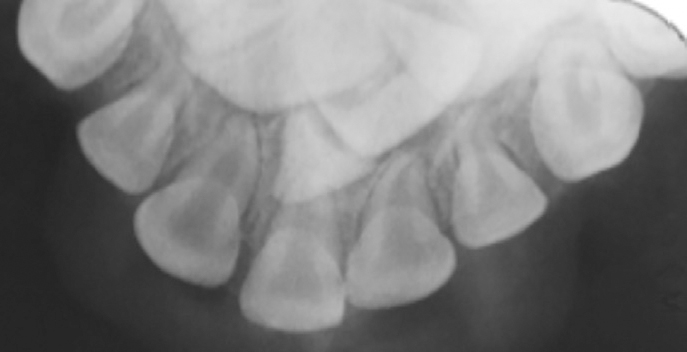
At 16 months old, a dental charting reported the eruption of his upper and lower primary incisors, together with an erupted symmetrical midline maxillary central incisor. By 3 years of age, all his other primary teeth had erupted and photographs and radiographs were taken (Figures 4 and 5).
Figure 4. Extra-oral view of male at 3 years old.Figure 5. Upper anterior occlusal radiograph of male at 3 years old.
The radiographic examination revealed that he also had a permanent supernumerary midline central incisor in addition to normal central and lateral incisors.
The upper anterior occlusal radiograph (Figure 5) showed:
Erupted URC, URB, URA, SMMCI, ULA, ULB, ULC;
Unerupted UR2, UR1, SMMCI, UL1, UL2.
An OPG was taken when he was 5 years old (Figure 6) which showed that all his permanent dentition was developing normally and there were midline supernumerary central incisors in both his primary and developing permanent dentitions.
Figure 6. Orthopantomogram of male at 5 years old.
When he was 4 years old, the Genetics Department had carried out a Deciphering Developmental Disorders (DDD) study which did not identify an explanation for the two siblings being born with SMMCI syndrome, but it was noted that it is not possible to examine every single region of genes. They did, however, identify that both children had an alteration in a gene called COLlagen type IV Alpha 2 chain (COL4A2), which was inherited from the maternal side. COL4A2 is a gene involved in the production of collagen and is important for collagen in the walls of blood vessels such as arteries.
Discussion
The presence of a symmetrical SMMCI of normal crown dimensions, situated precisely in the midline in both the primary and permanent dentitions, was first reported by Scott in 1958.4,5 The congenital absence of a maxillary central incisor is rare. Congenitally absent teeth in the primary dentition is very rare (<1%) and the presence of SMMCI is even more rare.6 If a primary tooth is absent, there will usually be an absence of the permanent tooth. The mechanism underlying the congenital missing maxillary incisor is unknown.1
Prompt recognition of SMMCI syndrome is vital as it can be a predicting sign of the more serious developmental defect HPE.2
The early diagnosis and therefore early treatment planning associated with SMMCI is of great importance as the dental anomaly may occur alongside other severe congenital malformations7 such as:
HPE cases with severe facial anomalies;
A microform in autosomal dominant HPE;
Other systematic abnormalities (including congenital heart disease, hypothyroidism or short stature/growth deficiency);
A feature of recognized syndromes (cleft lip and palate and ectodermal dysplasia);2
The diagnosis of SMMCI can be made prenatally at 18–22 weeks, or at birth, recognized by a midline prominence of the maxillary alveolus, an absent upper labial fraenum and an arch-shaped upper lip. A v-shaped palate is also characteristic due to a ridge in the midpalatal suture.8 Diagnosis should be made by 8 months old, with the eruption of a single symmetric maxillary central incisor.2
In 1992, Arliss and Ward recognized that congenital midline nasal malformation, such as choanal atresia, midnasal stenosis, and nasal pyriform aperture stenosis, are positively associated with SMMCI.9 Neonates are obligate nasal breathers for the first few months of life. SMMCI syndrome can often be recognized at birth as a result of life-threatening respiratory distress due to nasal obstruction.8 This association was slow to be recognized as these potentially life-threatening nasal airway obstructions are managed surgically in the immediate neonatal period, therefore months prior to the eruption of the tooth at 8 months of age.9
SMMCI was previously considered a simple midline defect of the dental lamina when Scott (in 1958) noted SMMCI as an isolated dental finding in a young girl. In 1968, Fulstow reported SMMCI associated with short stature, congenital heart disease, microcephaly and scoliosis in a young girl6 and, in 1976, the frequent association of short stature and solitary incisor was noted by Rappaport et al and they named the condition ‘monosuperoincisivodontic dwarfism’.5,9
SMMCI is now recognized as a possible predictor of holoprosencephalies of varying degrees.9 The significance of SMMCI lies in the fact that it may represent the mildest degree of manifestations in the spectrum of malformations seen in autosomal dominant HPE.1 Less severe forms of HPE can also present with mild facial dysmorphism, such as hypotelorism, iris coloboma and impaired midline cleavage of the embryonic forebrain (prosencephalon). In its most severe form (alobar HPE), there is no interhemispheric fissure and only a single eye (cyclopia), but this is unlikely to be seen by an orthodontist as the life expectancy is less.6
Less severely affected babies have normal or near normal cerebral development but may present with eye, nose and lip deformities, as well as cognitive developments and developmental delay.
Most cases with isolated SMMCIs will be part of the HPE spectrum.5 Atypical presentations of SMMCI may arise and the dental practitioner may be the first healthcare professional to recognize the condition. Medical liaison with a multidisciplinary input is essential, including geneticists, paediatricians and ENT, among others. Close liaison is especially important in the case of considering a joint GA for ENT and dental procedures. Management is to treat other systemic problems and the single tooth is mainly an aesthetic problem to be managed by combined orthodontics, prosthodontics and oral surgery.2
Becktor et al reported central incisor eruption within the normal age range, as expected, but suspected that transverse growth of the maxilla was restricted.10 Cho and Drummond looked at three cases with SMMCI and the treatment they required to correct their dental issues. All three patients did not have a labial frenum present, showed prominent midpalatal ridges and had an indistinct philtrum of the upper lip.6 The first child had a v-shaped maxillary arch and a unilateral crossbite on the left-hand side without mandibular shift. They were treated with orthodontic management alone. The second case had a hypoplastic midface, v-shaped maxillary arch, SMMCI and a unilateral crossbite on the right-hand side with mandibular shift. They were again managed by orthodontics alone. The third case reported the maxillary arch was u-shaped and the teeth were of acceptable alignment.
Conclusion
SMMCI should not be considered as a simple dental anomaly since it may be associated with more complex craniofacial malformations.6