Johns Hopkins University. Online Mendelian inheritance in man, OMIM. Number: 164210. 2022. https://tinyurl.com/msarr2mf (accessed June 2022)
Pruzansky S. Not all dwarfed mandibles are alike. Birth Defects. 1969; 1:120-128
Poswillo D. The aetiology and pathogenesis of craniofacial deformity. Development. 1988; 103:207-212
Poswillo D. Otomandibular deformity: pathogenesis as a guide to reconstruction. J Maxillofac Surg. 1974; 2:64-72
Caron CJ, Pluijmers BI, Wolvius EB Craniofacial and extracraniofacial anomalies in craniofacialmicrosomia: A multicenter study of 755 patients. J Craniomaxillofac Surg. 2017; 45:1302-1310
Elsten E, Caron C, Dunaway D Dental anomalies in craniofacial microsomia: a systematic review. Orthod Craniofac Res. 2020; 23:16-26
Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973; 35:302-328
Taysi K, Marsh J, Wise D. Familial hemifacial microsomia. Cleft Palate J. 1983; 20:47-53
Werler M, Sheehan J, Hayes C Demographic and reproductive factors associated with hemi-facial microsomia. Cleft Palate Craniofac J. 2004; 41:494-450
Kaban L, Moses M, Mulliken J. Correction of hemifacial microsomia in the growing child: a follow-up study. Cleft Palate J. 1986; 23:50-52
Steinbacher D, Gougoutas A, Bartlett S. An analysis of mandibular volume in hemifacial microsomia. Plast Reconstr Surg. 2011; 127:2407-2412
Vento A, LaBrie R, Mulliken J. The O.M.E.N.S. classification of hemifacial microsomia. Cleft Palate Craniofac J. 1991; 28:68-77
Gougoutas A, Singh D, Low D, Bartlett S. Hemifacial microsomia: clinical features and pictographic representations of the OMENS classification system. Plast Reconstr Surg. 2007; 120:112-120
Birgfeld C, Heike C, Saltzman B Reliable classification of facial phenotypic variation in craniofacial microsomia: a comparison of physical exam and photographs. Head Face Med. 2016; 12 https://doi.org/10.1186/s13005-016-0109-x
David D, Mahatumarat C, Cooter R. Hemifacial microsomia: a multisystem classification. Plast Reconstr. 1987; 80:525-535
Martelli-Junior H, Miranda RT, Fernandes CM Goldenhar syndrome: clinical features with orofacial emphasis. J Appl Oral Sci. 2010; 18:646-649
Goldenhar M. Associated malformations of eye and ear, particularly dermoid syndrome epibulbar-appendices, congenital auricular fistulas and its relations with manibulofacial dysostosis. J Genet Hum. 1952; 1:243-282
Grayson B, Boral S, Eisig S Unilateral craniofacial microsomia. Part I. Mandibular analysis. Am J Orthod. 1983; 84:225-230
Sharpen P. Neural crest and tooth morphogenesis. Adv Dent Res. 2001; 15:4-7
Seow W, Urban S, Vafaie N, Shusterman S. Morphometric analysis of the primary and permanent dentitions in hemifacial microsomia: a controlled study. J Dent Res. 1998; 77:27-38
Maruko E, Hayes C, Evans C Hypodontia in hemifacial microsomia. Cleft Palate Craniofac J. 2001; 38:15-19
Ongkosuwito E, de Gijt P, Wattel E Dental development in hemifacial microsomia. J Dental Res. 2010; 89:1368-1372
Craniofacial Microsomia: Aetiology, Classification and Clinical Features. Part 1 Clara Gibson Suhaym Mubeen Robert Evans Orthodontic Update 2025 15:3, 143-147.
‘This article gives an overview of craniofacial microsomia (CFM), its diagnostic features and relevant classification systems. Craniofacial microsomia is the most common facial anomaly after cleft lip and palate. It has a wide phenotypic variance and requires a multidisciplinary approach for comprehensive management. We outline both the facial and dental manifestations and orthodontic implications.
CPD/Clinical Relevance: CFM patients often require comprehensive dental and orthodontic care and it is necessary for the clinician to be aware of the clinical challenges in treating this cohort of patients. By having a thorough understanding of the aetiology and clinical features, it can help direct appropriate clinical care.
Article
Clara Gibson
Craniofacial microsomia (CFM) is a congenital facial condition that affects the structures derived from the first and second pharyngeal arches (OMIM #164210).1 It is an autosomal dominant condition with an estimated frequency of 1 in 3000–5000. CFM results in underdevelopment of facial structures, most commonly causing abnormal mandibular growth and morphology. Patients are affected to varying degrees with potential involvement of the ear, eye, nerves and soft tissues.2,3
Patients require longitudinal care of a multidisciplinary nature. Treatment is tailored to each individual patient on account of the variation in spectrum of issues related to the severity of each case. Treatment requires close interaction between medical and dental specialities including paediatric dentistry, restorative dentistry, orthodontics and oral and maxillofacial surgery. The general dental practitioner (GDP) plays a role in maintaining adequate oral health, which is a prerequisite for orthodontics and surgical correction, followed by definitive restorative and prosthetic dentistry where required.
Part 1 of this two-paper series outlines the aetiology of CFM and clinical presentation. Readers are directed to Part 2 for an overview of the management approaches (Orthod Update 2022; 15(4)).
Epidemiology
After cleft lip and palate, CFM is the most common craniofacial malformation, with the incidence reported as 1 in 3000 to 5000 live births.4 There is a slightly increased ratio of right-side incidence and in males (1.2:1).5
The condition is referred to by many other terms in the literature, including first and second branchial arch syndrome or oral–mandibular–auricular syndrome. Craniofacial microsomia is preferred to hemifacial microsomia, given that 10–16% of cases are bilateral.6,7
Aetiology
The exact aetiology is not yet fully known. Various theories have been proposed but owing to the wide phenotypic spectrum, it is postulated that there may not be one specific aetiological factor. Poswillo carried out experiments in mice using known teratogens and found a similar condition occurred due to a haematoma in the stapedial artery during in utero development.8 Family history may also play a role with studies demonstrating autosomal dominant and recessive patterns of inheritance.9 There is also evidence to link CFM with maternal risk factors such as smoking, diabetes and medications, but it is unlikely that these are the only aetiological factors.10 It is likely that a defect in neural crest cell migration is involved.
Classification system
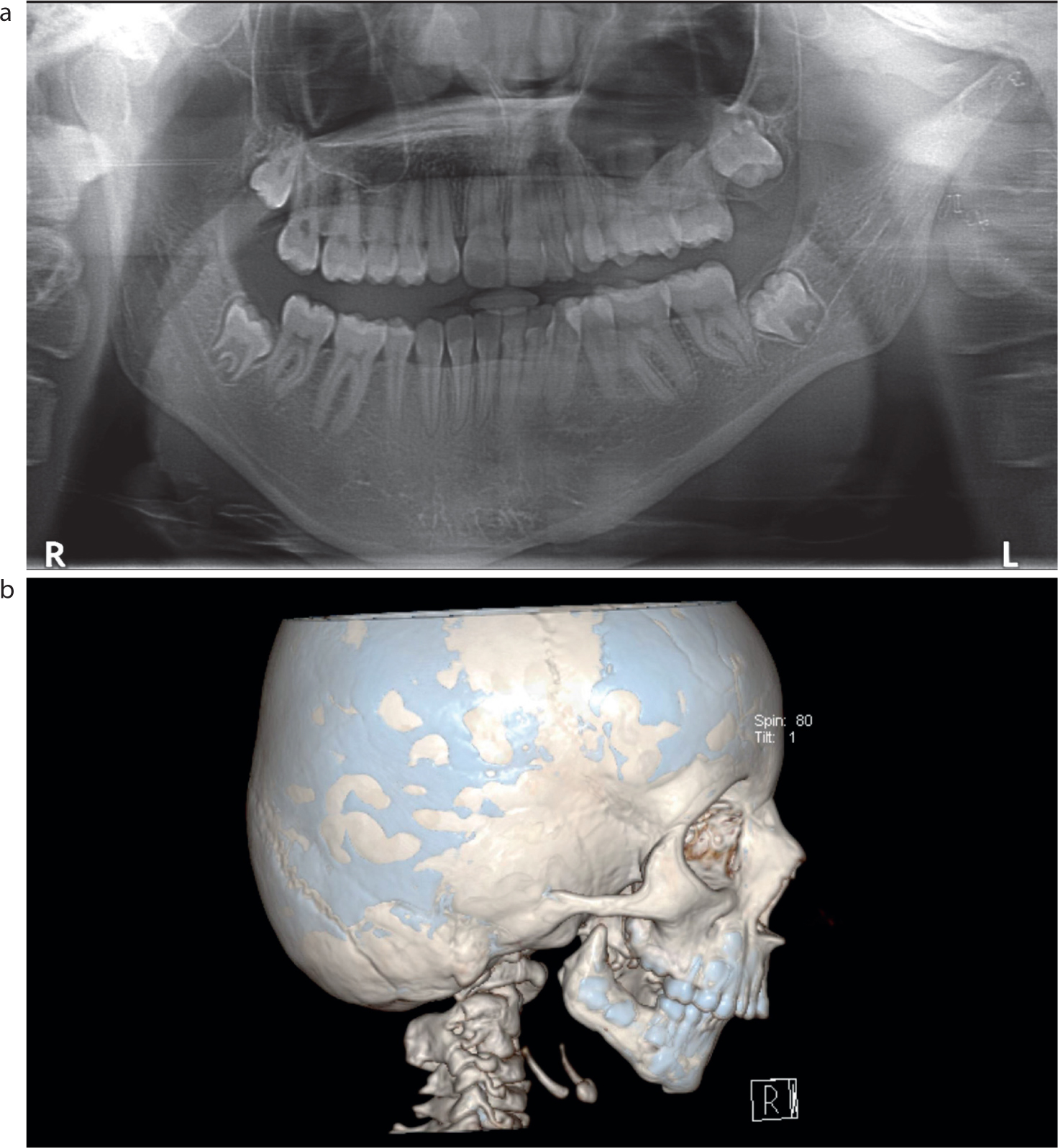
There are ranges of classification systems, which have evolved to represent the clinical presentation. The numerous systems highlight the challenges in classifying the condition due to broad phenotypic spectrum. Pruzansky used radiographic analysis of the size and shape of the mandible in a descriptive classification.2 This was later modified by Kaban et al, who subdivided category II into two groups on account of the extent of involvement of the glenoid fossa and whether surgical management would be required (Table 1).11 The Pruzansky classification is based on the structure of the proximal mandible on plain film radiographic analysis, but computed tomography imaging now provides a more accurate representation of the anatomical features and is routinely used in management of CFM cases (Figure 1).12
Pruzansky–Kaban category
Clinical features
Management
I
Hypoplastic mandible but normal in shape
Orthodontic alignment/functional appliance
May require orthognathic surgery to correct occlusion
II
IIa
Ramus, condyle and TMJ are present but abnormal in shape and hypoplastic. The glenoid fossa position is satisfactory
Does not require surgical treatment of TMJ. Manage as per I
IIb
Ramus is hypoplastic and abnormal in shape and position. TMJ is in abnormal positions (inferior, anterior, medial) with no condyle articulation with the temporal bone
Require reconstruction of TMJ with costochondral/iliac graft or total joint replacement with prosthesis
III
Absent ramus, condyle and TMJ
Requires surgical management. Costochondral or iliac graft or total joint replacement with prosthesis
Bimaxillary osteotomy likely, +/- distraction osteogenesis prior to this
Figure 1.
(a) Pruzansky IIa mandible viewed on OPG radiograph. (b) Pruzansky III mandible viewed with CT imaging.
The OMENS classification, superseded by OMENS+ to reflect extracranial manifestations, is in widespread use.13 OMENS is an acronym for orbit, mandible, ear, nerve and soft tissue with each of these components assigned a score from 0 to 3 in relation to their dysmorphic severity in that patient. In comparison to the mandibular-only system of Pruzansky and Kaban, the OMENS classification assesses the whole face and affected components. The OMENS classification can be used in its pictorial version14 (Table 2) and its use combined with clinical photographs (PAT-CFM) has been shown to be as useful as direct clinical examination of the patient.15 PAT-CFM is the Phenotypic Assessment Tool for Craniofacial Microsomia. This is the combination of the pictorial OMENS with a standardized set of facial photographs, in order to classify severity of CFM. It is used for multi-centre research projects, in lieu of patient examination.
The SAT scale (acronym for skeletal, auricular and soft tissue) has also been devised, to describe the mandibular, auricular and soft tissue deformities.16 This grading system is similar to the TNM-style multisystem classification that is used within the oncology field. The skeletal category has five levels of deformity (S1–S5), auricular category four levels (A0–A3) and soft tissue three levels (T1–T3). A patient with a significant deformity may therefore have a score of S5A3T3.
Clinical manifestations
General
CFM may be associated with other conditions, and patients with bilateral involvement have been found to have a more severe phenotype and extra craniofacial anomalies.5 Fifty-five percent of patients have extra-cranial anomalies, including vertebral fusion, kidney dysfunction and cardiac abnormalities.17 Goldenhar syndrome (also known as oculo-auriculo-vertebral spectrum) is believed to be a variant of CFM, with the additional features of epibulbar dermoids and vertebral anomalies.18
Facial
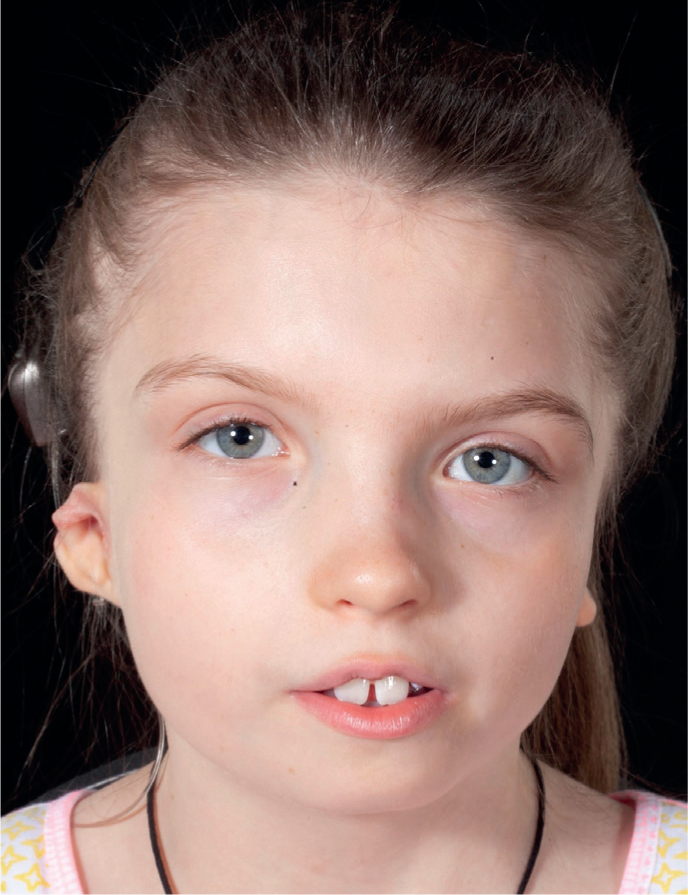
CFM causes hypoplasia of the facial skeletal and overlying soft tissue (Figure 2). The most important feature of craniofacial microsomia is facial asymmetry, which helps differentiate it from other craniofacial conditions such as Treacher Collins, in which both sides of the face are equally affected. There are no defined diagnostic criteria as the condition has a wide range of clinical presentations.
Figure 2.
(a) Skeletal and soft tissue hypoplasia viewed from frontal view; (b) profile left; (c) profile right. Note the asymmetry in tissue from each profile view.
A descriptive diagnosis is used, identifying the degree of hypoplasia or aplasia of any of the affected structures. The ear, maxilla, mandible, temporomandibular joint, facial musculature and fifth, seventh and twelfth cranial nerves may all be affected to varying degrees. Patients affected bilaterally may have structures affected to different degrees on each side (Figure 3). Ocular symptoms may range from slight malformation to complete anophthalmia (absence of an eye) and there may be microtia (underdevelopment of external ear), anotia (absence of external ear and narrowing or absence of ear canal) with or without impaired hearing. Pre-auricular skin tags are commonly found.
Figure 3. Bilateral CFM with left and right sides unequally affected.
Mandibular hypoplasia is the most prevalent anomaly (Figure 4). The mandible deviates upwards and towards the affected side, and a compensatory occlusal cant is common (Figure 5).19 The extent of mandibular involvement is described using various classification systems as shown in Table 1 and Table 2.
Figure 4.
(a, b) Mandibular hypoplasia.Figure 5. Compensatory occlusal cant.
Patients may have concurrent cleft lip and palate or macrostomia (enlargement of the mouth at the oral commissures). This is usually more pronounced on one side and so contributes to the appearance of marked facial asymmetry. There is often a soft tissue deficiency, due to concomitant oral/facial clefting, hypoplasia and deficiency of muscles, fat and skin.
Dental
There is a high incidence of dental anomalies in patients with CFM (Table 3).6 Dental tissue arises from the first pharyngeal arch, so this may be linked to the aetiology of defective neural crest cell migration.20 A systematic review found that delayed dental development is more frequently observed on the more affected CFM side.6 In addition, there was an increased incidence of four-cusp first molars, enamel defects, neonatal teeth, impactions and spacing.
Delayed dental development
Four-cusp first molars
Reduced mesio-distal dimensions of the mandibular molars
Neonatal teeth
Impacted teeth
Hypodontia
Enamel defects
Increased overjet
Restricted mouth opening
Research has shown that the mesio-distal dimensions of the mandibular molars on the affected side were smaller than control patients unaffected by CFM.21 In addition, the dimensions of the maxillary and mandibular molars on the ‘normal side’ in CFM patients were also smaller than controls, highlighting that CFM may well have bilateral effects, but to varying degrees. The incidence of hypodontia has been reported to be increased in CFM22 and the severity of hypodontia is correlated with how severely the mandible is affected, highlighting the relationship between development of the facial skeleton and teeth.22,23 The prevalence of missing teeth ranges from 6.7% to 33.3% although the pattern of hypodontia is similar to non-CFM patients with second premolars the most likely teeth to be absent (excluding third molars).6 A study has shown that Pruzansky–Kaban mandibles type IIb and III were associated with delayed dental development at a younger age (less than 5.5 years) when compared to mandibular type I, IIa and healthy controls.23 The team deduced that it is preferable to delay surgical treatment to a later age for patients type IIb and III – at least until after age 5.5 years – to allow for the later dental development.
Malocclusion is commonly found in CFM patients.6 The hypoplastic mandible may result in a class II malocclusion. The maxillary teeth may have over-erupted during growth to maintain a functional occlusion with the hypoplastic mandible, in particular if there are missing lower posterior teeth. Alternatively, the restricted vertical ramus growth on the affected side can lead to a concomitant restriction in vertical growth of the maxilla on the same side. The resultant cant can make full orthodontic correction without surgery a challenge. Patients with missing units may require prosthetic replacement; replacement of posterior units with implant prostheses is complicated by a potential lack of adequate bone.
Conclusion
The complex aetiology and variation in clinical presentation poses a challenge for both clinicians and patients alike. Owing to the heterogeneous nature of the condition, there are no clearly defined management protocols. This article is Part 1 of a two-part series and readers should refer to Part 2 for an overview of the management of patients with CFM.